Our recent review about studying enzymatic mechanisms using QM/MM mythologies has just been accepted on Israel Journal of Chemistry.
Abstract
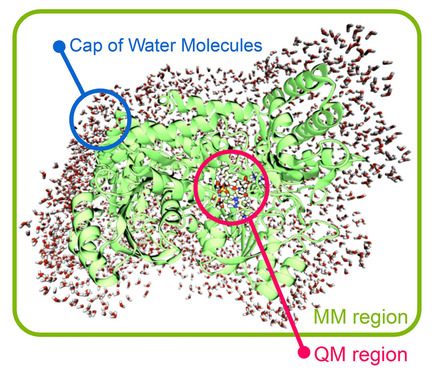
Quantum mechanics/molecular mechanics (QM/MM) methods are presently a well‐established alternative for the study of enzymatic reaction mechanisms. They enable the description of a small part of the enzyme, where reactions take place through QM, while the majority of the thousands of atoms that comprise these biomolecules are handled through MM. While different “flavors” and variations in the QM/MM field exist, this review will focus more on the application of the ONIOM methodology, presenting a fresh perspective on the application of this popular method in light of the growth in computational power and level of sophistication of the different methodologies that it can combine. In addition to a brief presentation of the basic principles behind these methods, this review will discuss different examples of applicability, common choices, practical considerations, and main problems involved, stemming from our experience in this field. Finally, a reflection on the future challenges for the next decade in the QM/MM modeling of enzymatic mechanisms is presented.
Authors: Magalhães RP, Fernandes HS, and Sousa SF